聚合酶鏈?zhǔn)椒磻?yīng)(Polymerase Chain Reaction,PCR)是體外合成雙鏈DNA的一種方法,其原理類(lèi)似于天然DNA的復(fù)制過(guò)程,其特異性主要依賴(lài)于與其目的片段兩端互補(bǔ)的特異引物和高特異性的酶。典型的PCR反應(yīng)有三個(gè)步驟:變性,退火,延伸,經(jīng)過(guò)多次循環(huán)反應(yīng),獲得目的片段。
試劑
DNA模板
特異性引物
dNTP Mix
PCR buffer
熱啟動(dòng)酶
操作步驟
1.配置反應(yīng)體系
將各成分依次加入到0.5ml的離心管中
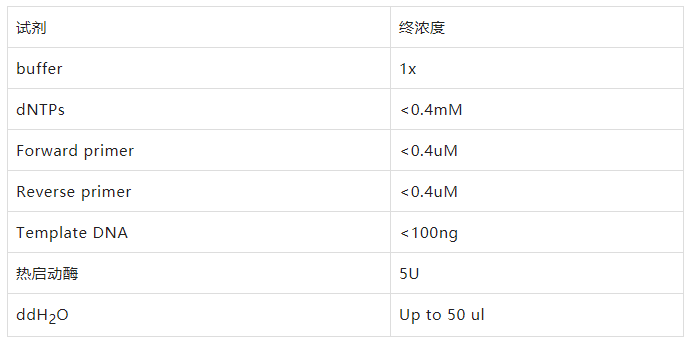
擴(kuò)增使用熱啟動(dòng)酶,可在常溫下操作,非熱啟動(dòng)型的酶要在低溫配置反應(yīng)體系。
2.按照試劑盒說(shuō)明書(shū)設(shè)置反應(yīng)時(shí)間和溫度,擴(kuò)增結(jié)束后電泳鑒定
3.瓊脂糖核酸電泳
(1)將電泳所用器具蒸餾水洗凈,架好梳子
(2)根據(jù)要分離的DNA片段大小制備合適濃度的瓊脂糖凝膠,準(zhǔn)確稱(chēng)取一定的瓊脂糖加入到錐形瓶中,加入30ml左右的電泳緩沖液(TAE或TBE)
(3)在微波爐中加熱融化后冷卻至40℃左右,充分混勻后倒入電泳槽中
(4)室溫下凝固40分鐘左右,小心拔出梳子,將凝膠放置電泳槽中準(zhǔn)備點(diǎn)樣
(5)在電泳槽中加入電泳緩沖液,漫過(guò)凝膠表面即可,點(diǎn)樣孔內(nèi)不要有氣泡
(6)樣品點(diǎn)樣前準(zhǔn)備好,(在八連管中)加入5ul樣品,1ul的6xLoding buffer和1ul的染料混合,用搶將混合后的樣品緩緩注入到點(diǎn)樣孔中,注意不要串孔
(7)按照正負(fù)極(紅正黑負(fù)),接通電源,電壓40-60V,時(shí)間30-40min,可根據(jù)溴酚藍(lán)的位置判斷是否終止電泳
(8)電泳結(jié)束,關(guān)閉電源,凝膠成像觀察,并對(duì)比marker確定片段大小
引物
引物是決定PCR反應(yīng)成敗的關(guān)鍵,要保證擴(kuò)增的準(zhǔn)確、高效,引物的設(shè)計(jì)要遵循如下原則:
1.引物的設(shè)計(jì)在cDNA的保守區(qū)域,在NCBI上搜索不同物種的同一基因,通過(guò)序列分析的軟件,得到不同基因相同的序列就是該基因的保守區(qū)
2.引物的長(zhǎng)度:15-28bp,長(zhǎng)度大于38bp,會(huì)使退火溫度升高,不利于普通Taq酶的擴(kuò)增
3.引物GC含量在40%-60%,Tm值最好接近72℃
4.因密碼子的簡(jiǎn)并性,引物的3’端最好避開(kāi)密碼子的第三位(最好為T(mén))
5.堿基分布錯(cuò)落有致,3’端不超過(guò)3個(gè)連續(xù)G或C
6.引物自身不要形成發(fā)夾結(jié)構(gòu),引物設(shè)計(jì)完成,要BLAST驗(yàn)證
模板
PCR擴(kuò)增對(duì)模板的要求不高,單鏈、雙鏈DNA均可作為擴(kuò)增片段,雖然PCR能夠擴(kuò)增微量的DNA,為了保證擴(kuò)增的特異性,對(duì)不同來(lái)源的模板,起始濃度有一定要求,如下:
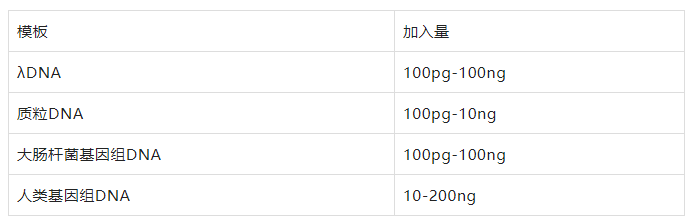
模板可以是純化后的樣品也可以是粗制品,不同來(lái)源的模板采取不同的處理方法,實(shí)驗(yàn)?zāi)0宓募兓胶?jiǎn)單越好,但模板中如果含有任何的Taq酶抑制劑、蛋白酶、核酸酶等,會(huì)干擾反應(yīng)。模板提取注意保存,不能放置太久,避免反復(fù)凍融并防止降解。多數(shù)人會(huì)忽略這一問(wèn)題,當(dāng)實(shí)驗(yàn)結(jié)果沒(méi)有任何條帶時(shí),可優(yōu)先考慮是否為模板降解的原因。
電泳緩沖液
1.50xTAE(pH8.5)
準(zhǔn)確稱(chēng)取Tris 242g,EDTA(Na)37.2g于1L燒杯中,加入800ml去離子水,攪拌溶解,接入57.1ml乙酸,充分?jǐn)嚢瑁{(diào)pH值8.5后定容至1L,室溫保存。每次使用時(shí)稀釋即可。
2.10xTBE(pH8.3)
準(zhǔn)確稱(chēng)取Tris 108g,EDTA 7.44g 硼酸55g于1L的燒杯中,加800ml去離子水,充分?jǐn)嚢枞芙猓{(diào)pH8.3后定容至1L,室溫保存。每次使用前稀釋即可。
上樣緩沖液
6xLoding buffer
0.25%二甲苯青,0.25%溴酚藍(lán),30%甘油溶于雙蒸水中,4℃保存。











